Cryoport Systems’ Temperature-Controlled Supply Chain Blog
Learn more from our expert leaders on the latest industry trends, global news, and company updates.

Supply Chain
04/29/2025
Optimizing Cryopreservation for Leukapheresis: Advancing Cell and Gene Therapy Supply Chains
The supply chain for fresh leukapheresis is one of the most complex logistical challenges in cell and gene therapy (CGT). As a critical starting material, leukapheresis must be collected from donors, processed, and transported within a limited time window. These challenges increase variability and risk, ultimately impacting the success of advanced therapies. Alexandre Michaux, Process Development & MSAT Manager at IntegriCell™ , recently presented his work on the development of an automated cryopreservation process for leukapheresis in a Poster Presentation at Advanced Therapies Congress in London.
Industry Insights
04/22/2025
Advancing Standardization in Cell and Gene Therapy Supply Chains
The future of cell and gene therapy depends not only on scientific advancements but also on the efficiency and reliability of its supply chain. In a recent panel discussion at Advanced Therapies Congress in London, Mark Flower, VP of Business Development, Cryopreservation at Cryoport Systems, explored the critical need for standardization, the challenges in achieving it, and the opportunities to improve logistics, manufacturing, and delivery systems.
Industry Insights
04/18/2025
Transforming Patient Access to CGT: Q&A with Aruna Mor
Aruna Mor, Chief Commercial Officer at Cryoport Systems, recently delivered a powerful keynote at Advanced Therapies Congress in London, shedding light on the complexities of patient access and the critical steps needed to overcome these barriers. We sat down with her to delve deeper into some of the key themes of her keynote, exploring the intersection of science, logistics, supply chain, regulation, and collaboration that will determine the future of Cell and Gene Therapy (CGT).
Industry Insights
04/14/2025
Enabling the Outcome Through Enhancing Patient Accessibility
Cell and Gene Therapy (CGT) has emerged as one of the most transformative areas in modern medicine, shaping the future of healthcare and changing the face of next-generation therapeutic approaches. Yet, despite the continued scientific progress, a critical challenge persists in ensuring these therapies are accessible to the patients who need them most. This challenge was the focus of the keynote address at the Advanced Therapies Congress in London, delivered by Aruna Mor, Chief Commercial Officer at Cryoport Systems, where she spoke about to the state of patient access within the field of advanced therapeutics.
Navigating Logistics
03/24/2025
Strengthening Global ATMP Logistics: How Cryoport Systems’ Netherlands Facility Supports Temperature-Controlled Supply Chains
The success of ATMPs depends on more than just scientific breakthroughs, it requires a highly controlled, risk-mitigated supply chain that ensures product integrity from research through commercialization. With cell and gene therapies moving across multiple clinical sites, manufacturing facilities, and regulatory jurisdictions, developers need a logistics partner that can support seamless, temperature-controlled supply chain management at a global scale.
Managing the Cold Chain
03/21/2025
Strengthening ATMP Development in Europe: How BioServices Centers in France Enable Efficiency and Scalability
As Advanced Therapy Medicinal Product (ATMP) developers progress from early-stage research to late-phase clinical trials and commercialization, biostorage, sample management, and regulatory-compliant logistics become increasingly complex. Efficiently managing these critical components is essential to ensure product integrity, regulatory compliance, and seamless supply chain operations.
Managing the Cold Chain
03/21/2025
Optimizing ATMP Cryopreservation: How IntegriCell™ Services in Belgium Ensure Cell Viability and Supply Chain Integrity
Cryopreservation is quickly becoming a critical yet often underestimated component of the Advanced Therapy Medicinal Product (ATMP) supply chain. The success of cell-based therapies depends on maintaining cell viability, stability, and consistency, yet variability in cryopreservation methods has compromised therapeutic outcomes.
Managing the Cold Chain
03/12/2025
Global Experts, Local Presence: Strengthening Europe’s Advanced Therapy Supply Chain
The development and commercialization of Advanced Therapy Medicinal Products (ATMPs) in Europe require a highly coordinated supply chain. From temperature-controlled logistics and biostorage to regulatory compliance, every stage of the ATMP lifecycle depends on precision, reliability, and risk mitigation
Navigating Logistics
03/12/2025
The Gateway to Europe: How Cryoport Systems’ Stevenage Supply Chain Hub Supports ATMP Development
For UK-based Advanced Therapy Medicinal Product (ATMP) developers, access to European clinical trial sites and commercial markets is a critical component of a successful growth strategy. Yet, navigating regulatory barriers, customs challenges, and supply chain complexities can create unnecessary delays, increase costs, and put the integrity of sensitive biologics at risk.
Navigating Logistics
03/04/2025
How Our Recent Innovative Solutions Meet Today’s Supply Chain Challenges
When advanced therapy organizations place innovation at the center of their approach to solving supply chain challenges, they prioritize the most important person involved – the waiting patient. An innovative, patient-centric approach reframes obstacles to focus on solutions that can ensure the vitality and protection of sensitive materials throughout every stage and phase of the journey to the patient’s bedside.
Industry Insights
02/12/2025
Ensuring Patient Impact in Advanced Therapy Logistics: A Conversation with Cryoport Systems’ Experts
The logistics behind advanced therapies aren’t just about moving shipments from point A to point B, they’re about protecting the integrity of life-saving treatments and ensuring that patients receive them without compromise. Following their discussion at Advanced Therapies Week 2025, we sat down with Aruna Mor, Chief Commercial Officer, and Mike Dybicz, Senior Vice President & Chief Product Development Officer, to dive deeper into how Cryoport Systems prioritizes patient impact at every step of the supply chain.
Patient Access and Awareness
02/12/2025
Advancing Cell and Gene Therapy on International Childhood Cancer Day
Every year on February 15th, International Childhood Cancer Day (ICCD) serves as a global platform to raise awareness about childhood cancer, advocate for improved access to care, and highlight the need for continued research and innovation. Pediatric cancer remains one of the leading causes of death among children worldwide, with approximately 400,000 new cases diagnosed each year.
Industry Insights
02/06/2025
The Patient Ferris Wheel: Centering the Patient in the CGT Supply Chain
In a recent interview at ATW, Cryoport Systems' leadership discussed how securing the future of medicine starts with minimizing risk and maximizing patient impact. Their conversation, The Patient Ferris Wheel – Centering the Patient in CGT Supply Chain, explored how Cryoport Systems is driving innovation in shipping systems designed specifically for advanced therapies.
Industry Insights
02/04/2025
Advancing Patient Outcomes Through Integrated Supply Chain Solutions
The landscape of advanced therapies is consistently shifting, requiring a seamless, high-quality supply chain to ensure the safe and effective delivery of life-saving treatments. In a recent interview at Advanced Therapies Week, Cryoport Systems leaders shared insights into how their integrated approach is shaping the future of supply chain management for the biopharmaceutical industry.
Patient Access and Awareness
02/04/2025
World Cancer Day: Advancing the Fight with Cell and Gene Therapies
Each year on World Cancer Day, the global community comes together to raise awareness, encourage prevention, and support those affected by cancer. It is a day of reflection, a time to recognize the immense progress that has been made in cancer research while acknowledging the work that still lies ahead.
Industry Insights
01/30/2025
Mapping the Future: Q&A with Mark Sawicki, Ph.D., on 2025 Trends in Cell and Gene Therapy
As the CGT industry enters a pivotal phase of commercial growth and therapy expansion, Cryoport Systems’ CEO, Mark Sawicki, Ph.D., shares his perspectives on where the market is headed.
Industry Insights
01/13/2025
Top 10 Industry Predictions for 2025
Cryoport Systems’ CEO, Mark Sawicki, provides a comprehensive analysis into the challenges and growth opportunities shaping the CGT industry.
Navigating Logistics
01/06/2025
The Future of Gene Therapy Logistics – A Pivotal Moment
Gene therapy is set to transform healthcare, but its success depends on logistics systems that can adapt and scale in line with the industry’s growth. Cryoport Systems is proud to support this exciting frontier with logistics solutions that prioritize product integrity, security, and compliance.
Navigating Logistics
12/30/2024
Supporting Compliance and Reducing Risk in Gene Therapy Logistics
When it comes to high stakes logistics in the gene therapy sector, adherence to regulatory standards isn’t just a box to check—it’s essential to delivering therapies that maintain integrity from production to patient. Cryoport Systems understands the unique risks in this space and has built compliance and risk mitigation into the very design of the Cryoport Elite™ Ultra Cold shipping system.
Navigating Logistics
12/23/2024
Meeting Market Needs through Collaboration and Customization
At Cryoport Systems, we understand that meaningful innovation comes not only from advanced technology but also from deeply collaborative relationships with our clients. Our collaborative approach has driven development of next-generation shipping systems and supply chain solutions, leaning on real-world insights from our clients to shape features, functionality, and future advancements.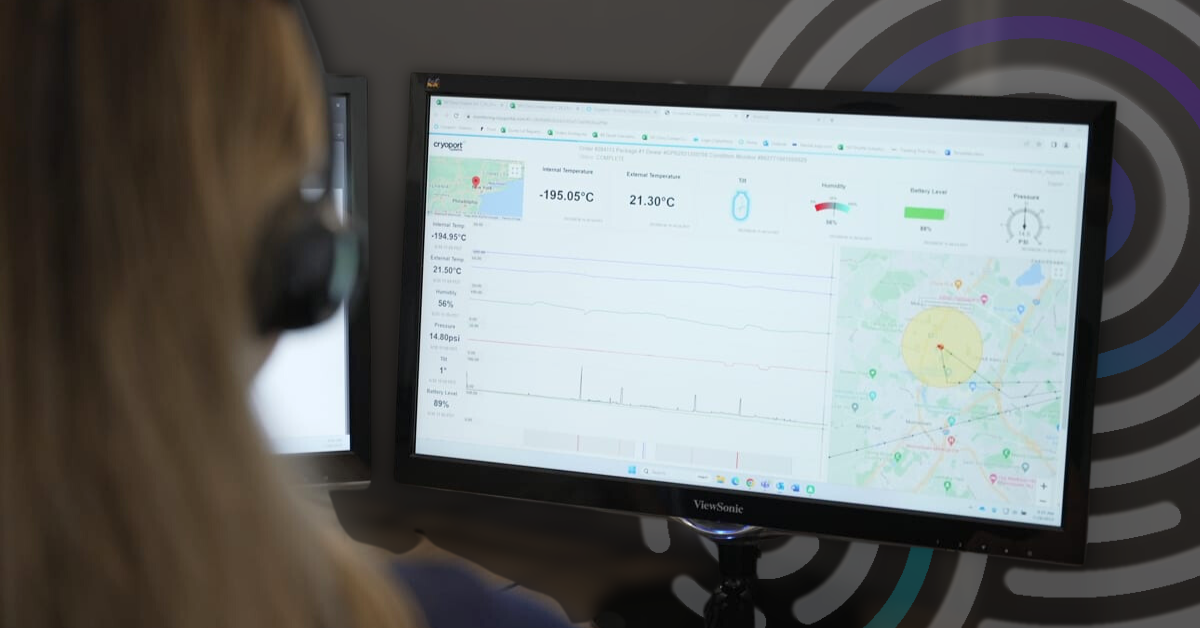
Navigating Logistics
12/16/2024
Solving Key Gene Therapy Logistics Challenges with Advanced Technology
Cryoport Systems developed the Cryoport Elite™ Ultra Cold shipping system with one mission in mind: to ensure that gene therapies reach their destination intact, uncompromised, and ready for patient treatment.
Industry Insights
12/11/2024
Reflecting on 2024: A Year of Groundbreaking Achievements
As we close out 2024, we reflect on a year marked by transformative milestones and significant growth at Cryoport Systems. This year, we took bold strides to redefine the future of supply chain solutions for life sciences.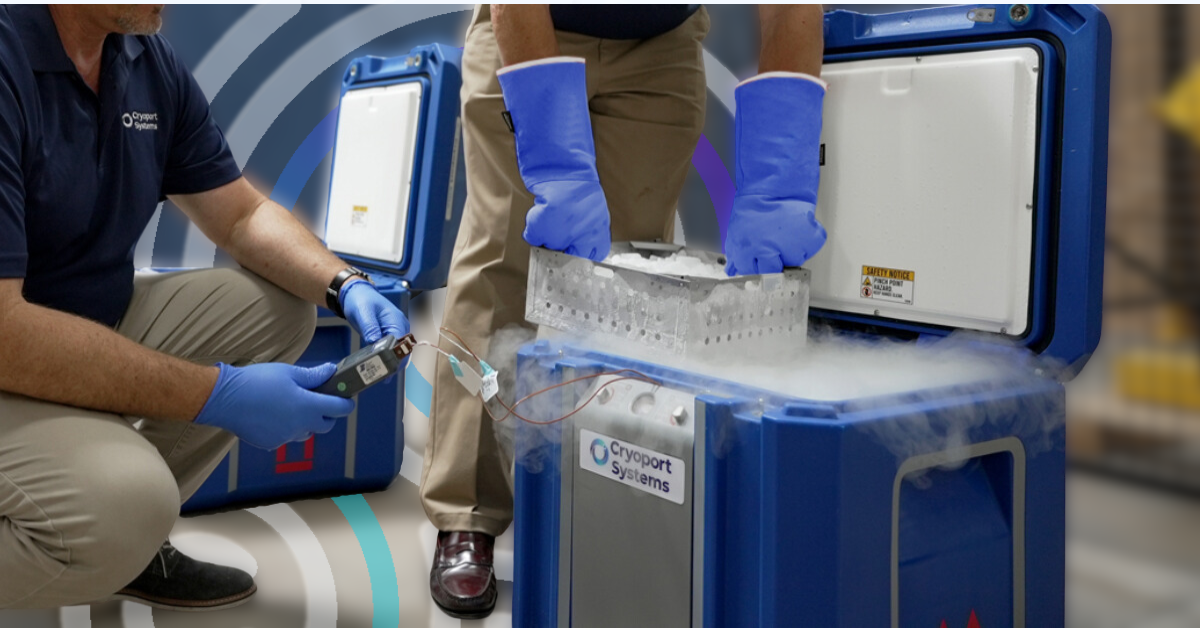
Navigating Logistics
12/09/2024
Meeting the Challenges of Gene Therapy Logistics with the Cryoport Elite™ Ultra Cold
As gene therapies move from research labs into clinical trials and commercialization, the need for secure, reliable logistics at scale is becoming an increasingly urgent need. Transporting these therapies requires a specialized approach.
Patient Access and Awareness
11/26/2024
National Family Health History Day: CGT Advancements for Generational Health
Since 2004, Thanksgiving Day in the U.S. has been paired with another lesser known but equally family-centric celebration – National Family Health History Day.
Navigating Logistics
10/04/2024
Certify Safety for Human-derived Materials with Advanced Therapy Shippers®
The risk of contamination can compromise life-saving therapies, making it a major concern within the world of transporting precision medicines.
Industry Insights
09/24/2024
Celebrating Our Recent Facility Expansion: A Recap of our Stevenage Global Supply Chain Hub Grand Opening Event
We recently had the pleasure of celebrating the official grand opening of our newest Global Supply Chain Hub in Stevenage, UK, alongside local clients and partners.
Navigating Logistics
09/03/2024
Cryoshuttle® Service Brings Local Support to Stevenage, England
In June, Cryoport Systems officially launched our newest Global Supply Chain Hub in Stevenage, England, a renowned center for cell and gene therapy development within the U.K.
Managing the Cold Chain
08/26/2024
3 Fundamental Services for Superior Master Cell Bank Management
The consistency of controlled conditions is vital throughout every step in producing advanced therapies, and the process usually begins with the secure handling and storage of the cells used for manufacturing.
Managing the Cold Chain
07/16/2024
IntegriCell™: Extending Cell Viability with Manufacture-ready Cryopreserved Leukopaks
During the COVID-19 pandemic, the restrictions on transportation and travel negatively impacted patients' access to life-saving treatments and support.
Managing the Cold Chain
06/27/2024
Cryoport Systems Celebrates Expanded French Facility with Ribbon Cutting Ceremony
Cryoport Systems commemorated the significant expansion of its Pont-du-Château facility in France with a ribbon cutting ceremony that officially launched the updated site.
Navigating Logistics
06/25/2024
The Cryoport Elite® Shipping System with Mike Dybicz, Cryoport Systems’ SVP & CPDO
In 2023, Cryoport Systems announced the launch of the Cryoport Elite™ shipping system.
Navigating Logistics
06/12/2024
Standardizing Compliance for Cell and Gene Transport with ISO 21973
It’s no secret that the cell and gene therapy market is one of the top-grossing markets within the life sciences industry.
Navigating Logistics
05/20/2024
Cryoport Elite® Shipping System: Industry-leading Advanced Therapy Support
The life sciences industry requires a robust shipping system that can handle the risks associated with transporting high-value commodities such as advanced therapies.
Managing the Cold Chain
04/16/2024
Overcoming Industry Obstacles for Cell and Gene Therapy Development
In the rapidly advancing world of cell and gene therapy (CGT) development, finding the proper solution to overcome common industry obstacles is a challenge that can impact entire production timelines.
Managing the Cold Chain
04/09/2024
National Holiday Facility Closures
As we look to the rest of 2024, Cryoport Systems wants to ensure that you’re up to date on our service schedule.
Navigating Logistics
03/27/2024
Tec4med: The Newest Addition to Cryoport’s Team
Cryoport Systems is a part of a nexus of supply chain solutions companies all under Cryoport, Inc. The other business units are CRYOGENE, CRYOPDP, and MVE Biological Solutions...
Industry Insights
02/22/2024
Examining 2023 & 2024: What’s to Come with President & CEO, Mark Sawicki
The cell and gene therapy industry is poised for many changes in 2024. With persistent challenges facing manufacturers and developers such as the retention of experienced talent or a general lack of funding for specialized therapies, the state of what’s to come feels more unsure than it has in recent years.
Managing the Cold Chain
02/06/2024
Enabling the Outcome with Cryoport Systems
Cryoport Systems entered the market over two decades ago to support the logistics processes of life sciences organizations. As industry demands evolved, our logistics capabilities expanded into a robust platform of supply chain management offerings.
Industry Insights
12/21/2023
Examining 2023 & 2024: Looking Back with President & CEO, Mark Sawicki
2023 was a year of highs and lows for the cell and gene therapy (CGT) market. The Alliance for Regenerative Medicine reported...
Managing the Cold Chain
11/14/2023
3 Services Your Biopharmaceutical Supply Chain Partner Should Offer
When it comes to choosing a cell and gene therapy shipping partner, not all providers are created equal.
Navigating Logistics
10/10/2023
The Crucial Role of Planning Ahead in Your Supply Chain Management
In the dynamic and ever-evolving realm of regenerative medicine and advanced therapies, the reliability of your supply chain could be the difference between success and failure.
Navigating Logistics
09/11/2023
4 Capabilities that Support a Superior Condition Monitoring Solution
For over 10 years, Cryoport Systems has been utilizing our near real-time condition monitoring solution that offers precise location information on shipments.
Navigating Logistics
08/16/2023
How Our Requalification Process Maintains Material Integrity
How can you be sure that your supply chain management partner understands the importance of cleanliness in standardized shipping practices?
Managing the Cold Chain
07/26/2023
A Glance into Our Houston & Morris Plains Global Supply Chain Centers
As cell and gene therapies continue to transform modern medicine, Cryoport Systems evolves its products and solutions to meet the needs of the industry and the patients it serves.
Managing the Cold Chain
05/31/2023
3 Services Your Biopharmaceutical Supply Chain Partner Should Offer
When it comes to choosing a cell and gene therapy shipping partner, not all providers are created equal.
Industry Insights
04/07/2023
Exclusive Q&A with Bruce McAfee, Director of BioServices Commercial Support
Cryoport Systems has extended its expertise in temperature-controlled supply chain solutions with vein-to-vein BioServices offerings.
Managing the Cold Chain
03/07/2023
Innovation and Risk Mitigation are Priorities for Cell&Co BioServices
At Cell&Co BioServices, a Cryoport Systems company, new product innovations are not adopted without significant testing. Recently our team dedicated 3 months to the creation and certification of a new type of cryogenic freezer technology.
Industry Insights
02/28/2023
State of the Industry: 2023 Predictions with Dr. Mark Sawicki
Cryoport Systems has grown to become a leader in the cell and gene therapy industry since its founding in 1999.
Industry Insights
12/13/2022
Documentary: Cell & Gene Therapy – Advancing The Next Generation of Pharmaceuticals
In this video documentary, American Pharmaceutical Review and Pharmaceutical Outsourcing magazine spoke with Cryoport Systems' CEO, Mark Sawicki...
Managing the Cold Chain
11/30/2022
Through Our Newly Launched Global Supply Chain Centers, Cryoport Systems Now Offers Kit Production
Cryoport combines its expertise in temperature-controlled supply chain solutions to develop a comprehensive offering of vein-to-vein services, one of which includes Kit Production.
Navigating Logistics
10/19/2022
Cryoport’s Chain of Compliance®: Collects, Interprets, and Leverages Comprehensive Data to Enable a Significantly Smarter Supply Chain
Companies vying to be the first to market with breakthrough treatments have a great deal riding on the success of their efforts.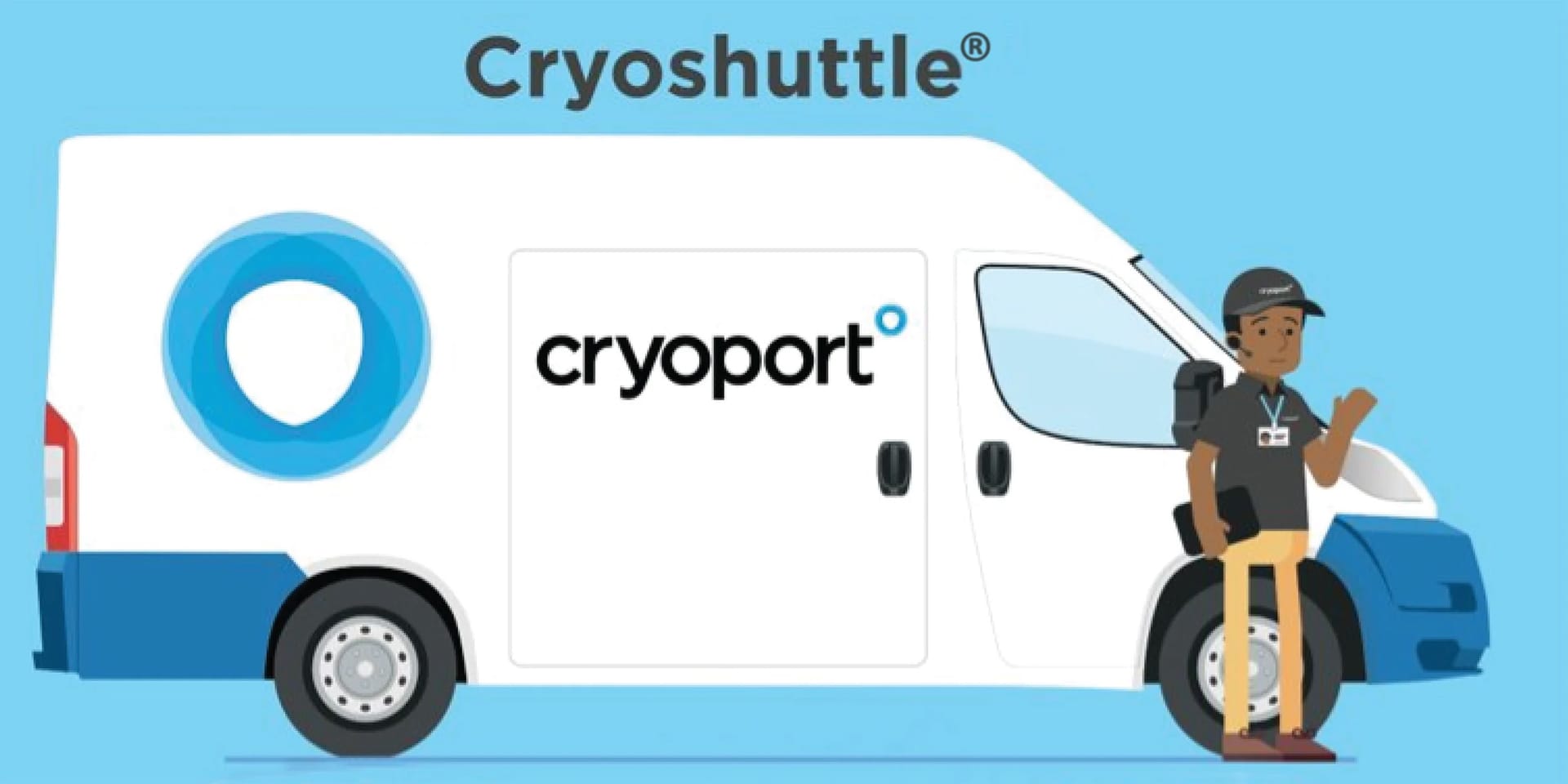
Navigating Logistics
10/13/2022
Cryoshuttle® Local Delivery and Pickup: Now Serving More Areas
Many biopharmaceutical companies, academic institutions, and manufacturing sites often need to move their valuable life-saving therapies and research materials locally for a variety of reasons...
Navigating Logistics
08/29/2022
The Cryoport Way: A Leap Forward in Standardizing the Regenerative Medicine Supply Chain
As cell and gene therapies are rapidly moving beyond clinical trials and into commercial markets, the safe and effective delivery of these life-saving treatments needs to be a priority for advanced therapy developers.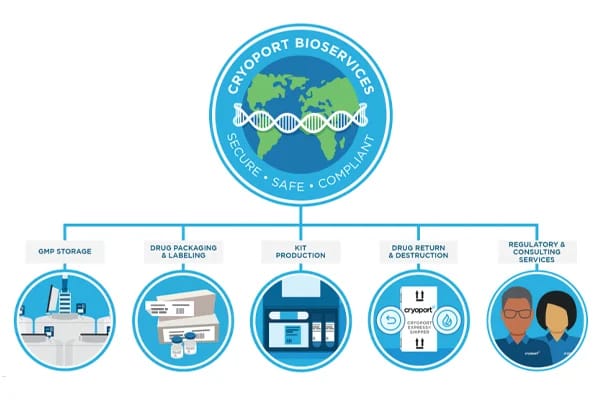
Managing the Cold Chain
08/22/2022
Cryoport Systems’ Global BioServices: Secondary Packaging and Labeling Solutions
Cryoport Systems’ highly anticipated Global Supply Chain Centers have been operational for a few months, and customers continue to move biopharmaceutical materials into our Houston, Texas...
Patient Access and Awareness
07/20/2022
The Untapped Potential for Cord Blood as Allogeneic Starting Material
In support of Cord Blood Awareness Month, Cryoport Systems’ partner, Be The Match BioTherapies®, led a webinar focused on the untapped potential and benefits of cord blood for prospective therapies.
Managing the Cold Chain
06/06/2022
Cryoport Systems Global Supply Chain Center: Ready for Launch!
We are quickly approaching the Grand Opening of our first two Global Supply Chain Centers in Houston, TX on June 14th and Morris Plains, NJ on June 16th, 2022.
Navigating Logistics
05/19/2022
New Product Launch: The Latest Enhancements to Cryoport Express® Standard & Combo Cryogenic Shippers
At Cryoport Systems, we are continuously striving to align with the latest science and technology advancements in our products and services, integrating the most advanced packaging, informatics...
Industry Insights
02/11/2022
Are You Ready for the Upcoming Data Network Transition? Here’s Everything You Need to Know
The future for global advanced therapy shipments requiring critical near real-time chain of custody, chain of condition and Chain of Compliance® reporting will soon face a significant technology challenge.Categories
- All (58)
- Industry Insights (16)
- Managing the Cold Chain (16)
- Navigating Logistics (21)
- Patient Access and Awareness (4)